浙江国内注册eCTD服务放心可靠
美国药物主文件(Drug Master File, DMF)是向FDA提交的机密技术文件,用于支持药品生产、质量控制及合规性审查。以下为申报的要点和流程总结: DMF概述与类型 ?定义与作用 DMF是药品生产全过程的详细档案,包含原料药、辅料、包装材料等的生产设施、工艺、质量控制等信息,供制剂厂商引用以支持其注册申请。其意义在于保护企业机密的同时,满足FDA对供应链透明度的要求。 ?DMF类型 ?Ⅱ类:原料药、中间体及制剂(如微生物外泌体、细胞株等生物制品均属此类)。 ?Ⅲ类:包装材料。 ?Ⅳ类:辅料、着色剂等添加剂。 ?Ⅴ类:非临床/临床数据等特殊信息(需FDA预先批准)。 注:Ⅰ型(生产设施与人员)已于2000年停用。欧盟eCTD注册外包相关技术支持。浙江国内注册eCTD服务放心可靠

eCTD生命周期管理与变更提交:欧盟要求eCTD申报资料覆盖药品全生命周期,包括提交、补充申请及实质性变更。例如,增成员国需提交“附加成员国序列”,审评时间约52-83天;重大变更(如生产工艺调整)需创建序列并通过CTIS平台更模块3和模块1的GMP证明。技术验证工具(如EDQM推荐的检查软件)需在每次提交前运行,确保XML骨架文件与PDF书签层级符合规范。此外,电子签章需符合《欧盟电子签名法》,并在模块1中明确标注法律效力。欧洲通用提交门户(Common European Submission Portal,CESP)是欧盟及成员国药品监管机构间用于电子化提交申报资料的重要平台。以下是关于CESP的详细介绍: CESP是由欧盟药品监管部门负责人网络(HMA)合作开发的在线交付系统,旨在为药品注册申请者、利益相关方和监管机构之间提供统一、安全的电子提交通道。其设计初衷是简化跨国申报流程,允许通过单一门户向多个欧洲国家的药监部门同时提交申请,避免了重复操作。杨浦区药品注册eCTD供应商加拿大ANDA注册申报相关技术支持。
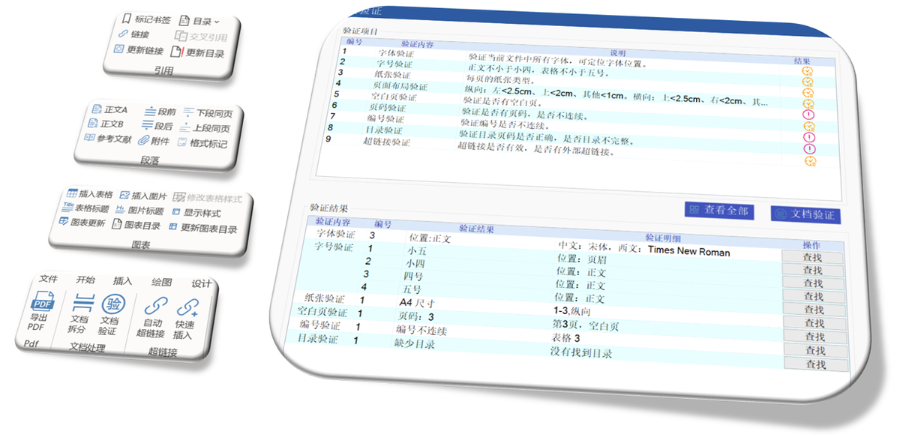
多国审评程序与eCTD递交途径的适配:欧盟药品审评程序包括集中(CP)、分散(DCP)、互认(MRP)和国家程序(NP),eCTD需适配不同程序的递交要求。例如: ?集中审评程序(CP)?:通过EMA的eSubmission Gateway提交,审评时限约240个工作日,eCTD需包含完整的模块1-5及多语言标签文件。 ?分散审评程序(DCP)?:需通过CESP(欧盟共同提交门户)递交,参考成员国(RMS)主导审评,eCTD需支持多国同步评估的模块化拆分。 ?互认程序(MRP)?:已授权成员国作为RMS,eCTD需包含基线序列(Baseline Sequence 0000)以整合历史审评数据,并通过CMDh协调分歧。
澳大利亚的药品电子通用技术文档(eCTD)注册申报体系是澳大利亚y药品商品管理局(TGA)推动药品审评现代化的重要举措。eCTD作为国际通行的电子化注册申报标准,通过结构化数据格式(如XML)整合了药品质量、安全性和有效性的技术文档,实现了从传统纸质递交向数字化流程的转型。根据TGA要求,eCTD需遵循通用技术文档(CTD)框架,分为五个模块:模块1包含澳洲特定的行政信息(如产品说明书草案和GMP证明);模块2为质量、非临床及临床研究的综述与总结;模块3至模块5则分别涵盖药学、非临床和临床的详细数据。澳大利亚自2024年起加速推进eCTD实施,要求创新药注册申报优先采用该格式,以提升审评效率并支持全球同步申报。 申报流程上,企业需通过TGA指定的电子提交门户(如eSubmission Gateway)上传eCTD序列,并在受理后5个工作日内同步提交纸质版模块1-5资料。欧盟IND注册申报相关技术支持。
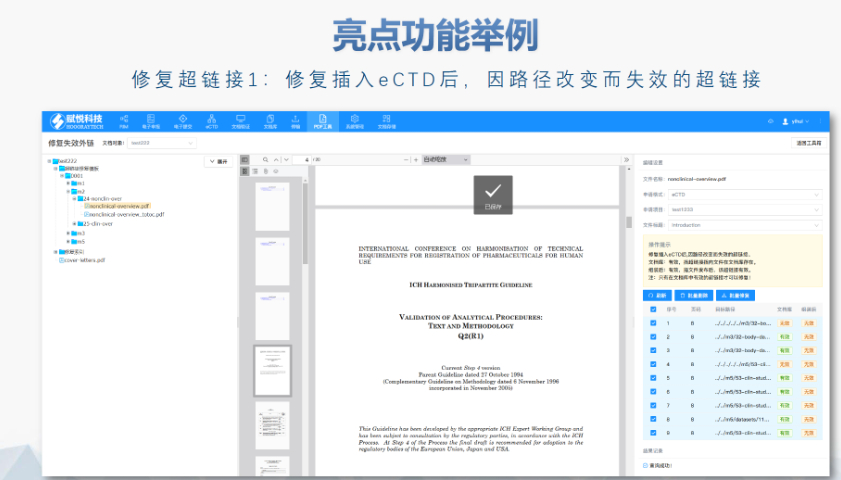
eCTD的法规框架与技术规范:欧盟eCTD的法规层级包括指南(Guidelines)、指令(Directive)和法规(Regulation)。其中,法规(如CTR)具有直接法律效力,而指南(如ICH eCTD规范)则为技术操作提供参考。eCTD的结构需符合欧盟模块1规范(DTD 3.0+),包含行政文件(模块1)、质量数据(模块3)及临床研究报告(模块5)等内容,并通过XML文件实现数据互联。例如,CEP(欧洲药典适用性证书)的eCTD申报需单独构建信封(Envelope)和模块1,并指定标识符(UUID)以确保技术合规性。中DMF注册申报相关技术支持。江苏中国eCTD发布软件
美国NDA注册申报相关技术支持。浙江国内注册eCTD服务放心可靠
电子签章与安全性 FDA要求所有PDF文件需经数字签名,并通过MD5校验确保传输完整性。签章需符合21 CFR Part 11的电子记录规范,部分情况下允许临时放宽(如期间的远程签署)。 ?多模块协同验证 模块1(行政文件)的区域性元数据(如申请类型、联系人信息)需与模块2-5的内容逻辑一致。例如,生物制品的3.2.R扩展节点命名需遵循特定规则,而化学药品则禁止使用此类扩展。 ?验证工具与流程 主流工具如LORENZ eValidator支持自动化验证,生成包含错误定位与修复建议的详细报告。企业需在提交前完成内部验证,并通过“药品业务应用系统”推送受理状态。 ?常见问题与规避策略 高频错误包括PDF安全设置、书签链接失效、STF(研究标签文件)缺失等。例如,未在5.3.1章节标注研究ID会导致验证警告,需通过说明函解释。企业可通过建立标准化模板库和预检流程降低风险。 ?后续监管与更 FDA定期更验证标准(如2022年增临床试验数据完整性检查),企业需通过订阅官方通知或第三方服务商获取动态浙江国内注册eCTD服务放心可靠
- 高新区中国eCTD欢迎选购 2025-05-14
- 静安区生物制品eCTD使用 2025-05-14
- 芜湖新药eCTD是什么 2025-05-14
- 吴江区赋悦科技eCTD供应商 2025-05-14
- 南京生物制品eCTD注册系统 2025-05-14
- 上海化学药品eCTD格式 2025-05-09
- 南京电子申报eCTD哪个品牌好 2025-05-09
- 太仓NDAeCTD服务价格 2025-05-09
- 浦东新区原料药eCTD文件如何制作 2025-04-26
- 南京新药eCTD找哪家 2025-04-26
- 湖南巡检养老机器人定制 2025-06-07
- 杭州消费电子类线束 2025-06-07
- 家校互联视频话机厂家批发 2025-06-07
- 淮安特殊光解膜厂家直销 2025-06-07
- 湖北正版ANSYS HFSS 2025-06-07
- 重型智能营销产品 2025-06-07
- 浙江RFID通道门销售公司 2025-06-07
- 无锡好的热塑性复合材料量大从优 2025-06-07
- 经销商营销获客方案订制 2025-06-07
- 河北工业机器人力控搬运 2025-06-07