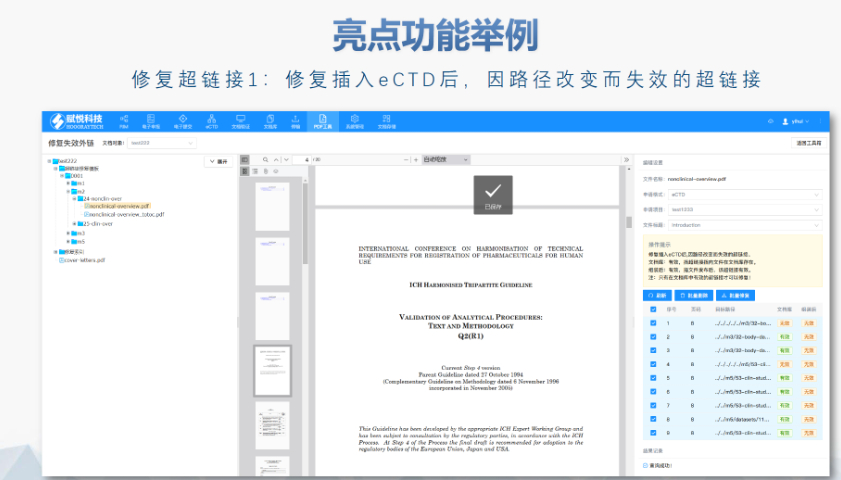
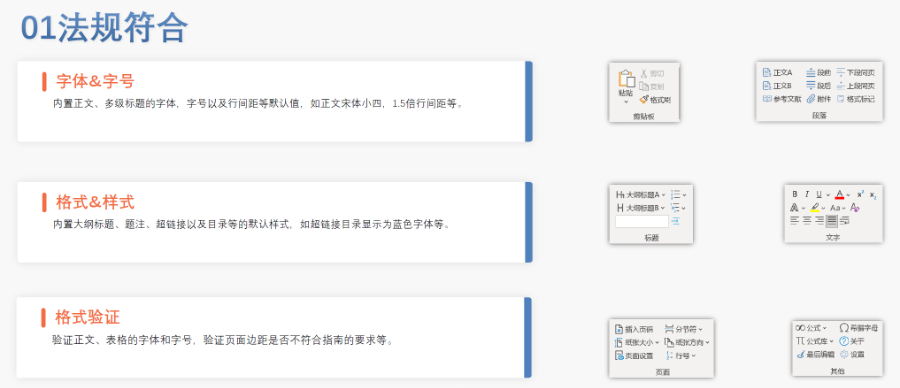
吴江区CDE eCTD常用解决方案
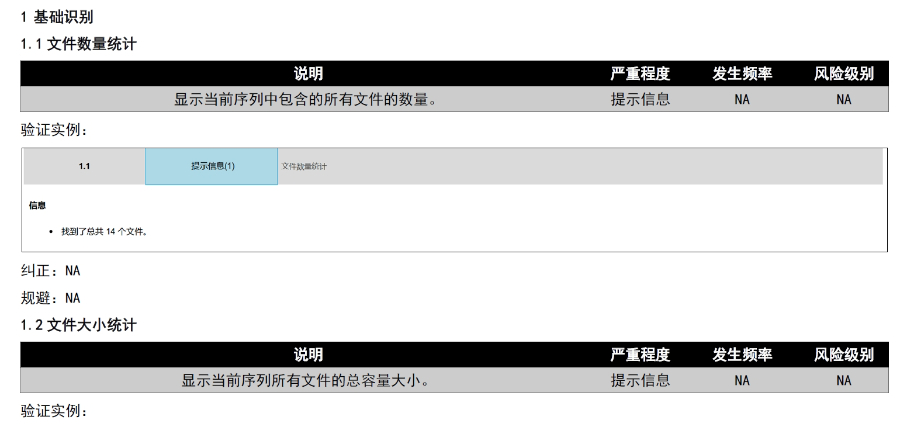
eCTD验证标准的严格性与分类:欧盟对eCTD的验证要求分为“错误”“警告”和“提示信息”三级,其中“错误”项直接导致申报被拒。验证项目涵盖六大类共149条,包括文件命名规范(如路径长度限制)、PDF可读性(禁止密码保护)、XML骨架文件完整性等。例如,文件扩展名必须符合规范(如.xpt用于临床数据集),而文件夹层级需避免空目录或混合存放文件。相较于中国《电子申报验证标准》的简化版(54条),欧盟的验证体系更为复杂,体现了其高标准的技术监管。eCTD验证标准相关技术支持。吴江区CDE eCTD常用解决方案
申报流程与要求 ?资料准备 ?内容要求:包括产品描述、生产工艺(原材料来源、设备参数等)、质量控制标准(SOP、稳定性数据)、安全性与毒性研究等。 ?格式规范: 采用CTD(通用技术文件)格式,按模块分章节(如模块3为CMC数据)。 电子提交需符合eCTD标准(文件小于10GB通过ESG系统提交,超过可选用CD-ROM)。 ?提交与注册 ?预分配DMF号:需在提交前申请,确保文件与编号绑定。 ?授权书(LOA)?:需向引用DMF的制剂厂商提供授权信,明确可查阅的章节。 ?费用:Ⅱ类原料药DMF需缴纳年费(2024年约9,468美元)。 ?FDA审核流程 ?行政审评:2-3周内确认文件完整性。 ?完整性审评(CA)?:针对Ⅱ类DMF,约60天。 ?技术审评:在DMF被制剂申请(如ANDA、NDA)引用时启动,周期60-180天。 ?结果反馈:FDA可能要求补充数据,但DMF本身无“批准”状态,通过后可能收到“无进一步意见函”(No Further Comment Letter)。太仓国产eCTD常用解决方案加拿大ANDA注册申报相关技术支持。
欧盟eCTD的递交途径与技术要求 不同审评程序对应不同递交渠道:集中程序(CP)通过EMA的eSubmission Gateway或Web Client提交,分散程序(DCP)和互认程序(MRP)则需使用欧盟通用提交门户(CESP)。文件结构需严格遵循模块化要求,例如CEP申请需包含模块1(行政文件)、模块2(质量概述)和模块3(技术文档),且XML主干文件须符合EDQM的特定命名规则。此外,所有PDF文件需无密码保护、可全文检索,并嵌入层级书签以支持快速审阅。 CEP申请的eCTD递交特殊性 CEP程序自2018年起强制采用eCTD格式,重点评估原料药是否符合欧洲药典标准。其模块1需包含EDQM申请表、简历及变更说明表,模块2需使用EDQM提供的质量概述模板,模块3则按CTD格式组织3.2.S章节内容。CEP与ASMF(活性物质主文件)的主要区别在于性:CEP无需关联上市许可,且审评由EDQM完成。
仿制药作为提高药物可及性与可负担性的一类药物,2012年以前,注册审评是不收取任何费用的,但当时仿制药申请积压严重,从申报到获批需要3~5年的时间。 美国国会于2012年颁布了仿制药使用者费用修正案(Generic Drug User Fee Amendments, GDUFA),该法律要求制药行业支付一定的用户费用,以补充仿制药申请的审评以及现场检查的费用,减少仿制药申请积压,缩短审评时间,增加基于风险的现场检查等,其目的是加快公众获得安全有效的仿制药,并降低行业成本。 GDUFA必须每五年重授权一次,于2017年更(GDUFA II),于2022年更(GDUFA III); 目前收费种类分为以下四种:ANDA审评费、DMF审评费,在审评时一次性缴纳;项目费(Program fee)、设施费(Facility fee),是上市后每年缴纳一次。澳大利亚eCTD注册外包相关技术支持。
争议解决与法律救济 若申请人对审评结果有异议,可向EMA的CHMP申请重审查,或在欧盟法院提起行政诉讼。eCTD的完整提交记录可作为法律证据,证明申请人已履行合规义务。EDQM设立仲裁委员会,处理CEP程序中的技术争议。 行业趋势与竞争格局 全球eCTD服务市场年增长率达12%,欧盟占据35%份额,主要服务商包括PharmaLex、Certara等。头部药企通过自建IT团队降低成本,而中小型企业依赖外包以专注研发。人工智能(AI)在文件自动生成和审评意见预测中的应用逐渐增多。 患者参与与透明度提升 EMA通过公开eCTD摘要(如模块2.5临床概要)增强审评透明度,患者组织可提交意见影响审评决策。部分成员国要求模块1包含患者语言版本说明书,以提升用药依从性。未来,eCTD4.0或支持直接链接患者反馈平台,实现全生命周期互动。美国API的DMF申报相关技术支持。吴江区CDE eCTD常用解决方案
加拿大DMF注册申报关技术支持。吴江区CDE eCTD常用解决方案
eCTD 4.0版本的过渡与升级:FDA于2023年启动eCTD 4.0技术试点,2024年9月正式接收申请,计划2029年完成全过渡。4.0版本改用HL7 RPS标准替代XML,支持双向通信和跨申请文件复用,例如同一Study ID可在IND和NDA享。模块1的校验码从MD5升级为SHA-256,主干文件由改为,序列号取消前导零(如“1”而非“0001”)。企业需同步更软件系统以适应架构。DMF与IND申报的特殊要求:针对Type II(原料药)和Type IV(辅料)DMF,eCTD模块3需详细描述生产工艺、稳定性数据,并附分析证书(COA)。FDA要求DMF持有人指定美国境内代理人,确保沟通效率,且LOA(授权书)需明确引用范围。IND安全性报告(如SUSAR)需通过eCTD模块5.3.5提交,15天内完成,并嵌入CIOMS或MedWatch表格。2024年指南强调,临床数据库需以SAS XPORT格式提交,单个文件超过4GB需拆分并说明规则。吴江区CDE eCTD常用解决方案
- 高新区中国eCTD欢迎选购 2025-05-14
- 静安区生物制品eCTD使用 2025-05-14
- 芜湖新药eCTD是什么 2025-05-14
- 吴江区赋悦科技eCTD供应商 2025-05-14
- 南京生物制品eCTD注册系统 2025-05-14
- 上海化学药品eCTD格式 2025-05-09
- 南京电子申报eCTD哪个品牌好 2025-05-09
- 太仓NDAeCTD服务价格 2025-05-09
- 浦东新区原料药eCTD文件如何制作 2025-04-26
- 南京新药eCTD找哪家 2025-04-26
- 松江区新型的SMT贴片加工OEM代工 2025-06-02
- 天津新一代智慧博物馆 2025-06-02
- 广东星闪模块有优势 2025-06-02
- 普陀区通信网络安全防护咨询 2025-06-02
- 中国香港怎样企业租赁 2025-06-02
- 安徽景区智慧博物馆开发公司 2025-06-02
- 辽宁有名DC380卡片打印机销售电话 2025-06-02
- 数字农田erp哪个好 2025-06-02
- 北京vr虚拟拍摄 2025-06-02
- 北京通用工控一体机厂家直销 2025-06-02